FUNCTIONAL RELEVANCE OF PROTEINS’ STRUCTURAL DYNAMICS
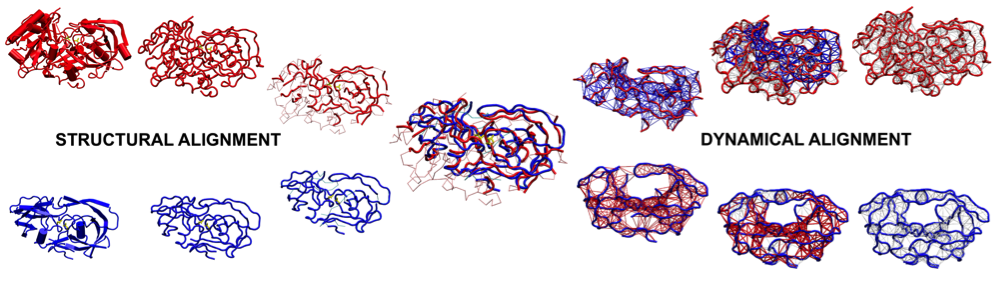
Proteins have evolved not only under the pressure to fold rapidly and reversibly into a well-defined overall shape but
also for being capable of harnessing thermal energy toward realizing specific structural fluctuations that are
functionally oriented. Accordingly, our current understanding of proteins’ function emphasizes the dynamic nature of
these biopolymers. Rather than focusing on a single molecular structure, we envision the native state as an ensemble
encompassing many distinct structural states. It is the interconversion between these states that enables proteins to
change their physico-chemical properties in a controlled fashion, examples of such interconversions are: the open and
closed forms of the pore of an ion channel and the bound and unbound forms of an enzyme-substrate complex. Thus,
understanding the function of a protein entails, in essence, studying its structural fluctuations and how these change
in response to a change in some environment factor, e.g.transmembrane potential, pH, temperature etc. Our research is
directed toward the development of methods to effectively describe the structural dynamics of proteins and to perform
comparisons within and across distinct protein families.
L. Ponzoni, G. Polles, V. Carnevale, and C. Micheletti. SPECTRUS: a dimensionality reduction approach for identifying
dynamical domains in protein complexes from limited structural datasets. Structure, 23:8, 1516-1525, 2015.
A. Zen, V. Carnevale, A. M. Lesk, and C. Micheletti. Correspondences between low-energy modes in enzymes: Dynamics-based
alignment of enzymatic functional families. PROTEIN SCIENCE 17, 918 (2008).
V. Carnevale, F. Pontiggia, and C. Micheletti. Structural and dynamical alignment of enzymes with partial structural
similarity. JOURNAL OF PHYSICS-CONDENSED MATTER 19 (2007).
V. Carnevale, S. Raugei, C. Micheletti, and P. Carloni. Convergent dynamics in the protease enzymatic superfamily.
JOURNAL OF THE AMERICAN CHEMICAL SOCIETY 128, 9766 (2006).
V. Carnevale, C. Micheletti, F. Pontiggia and R. Potestio Bridging the Atomic and Coarse-Grained Descriptions of
Collective Motions in Proteins. In A. Kolinski editor, Multiscale Approaches to Protein Modeling, Springer Verlag,
2010.