Daniele Granata, Luca Ponzoni, Cristian Micheletti, and Vincenzo Carnevale
PNAS, 114(50), E10612-E10621, 2017
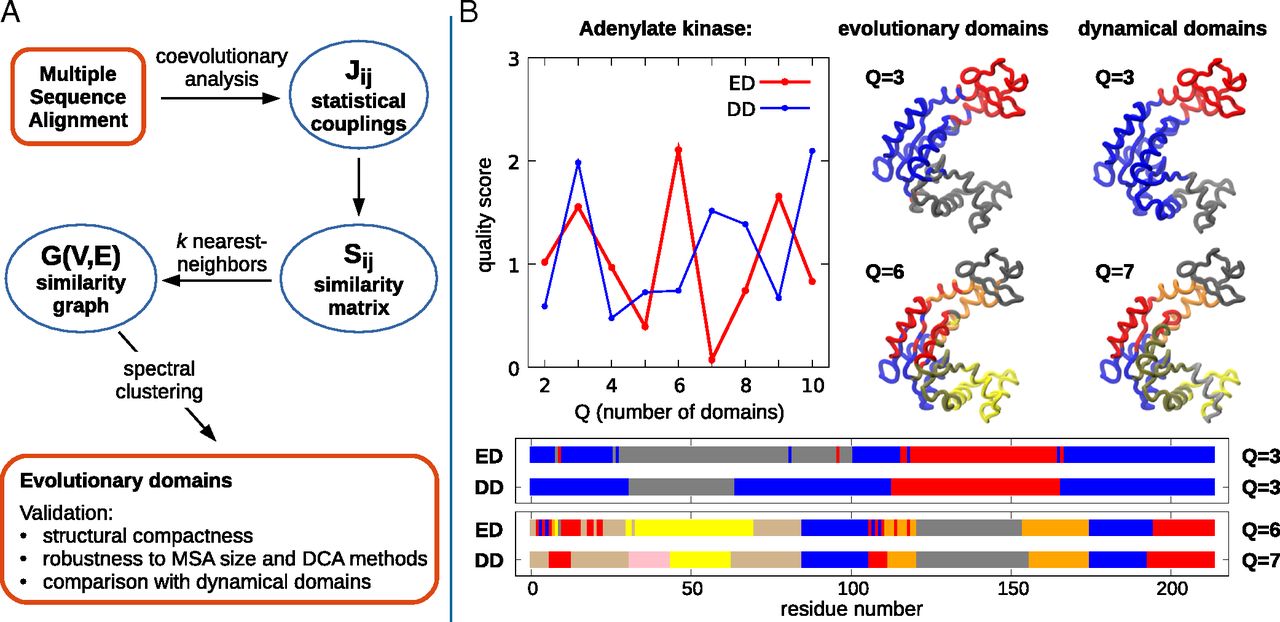
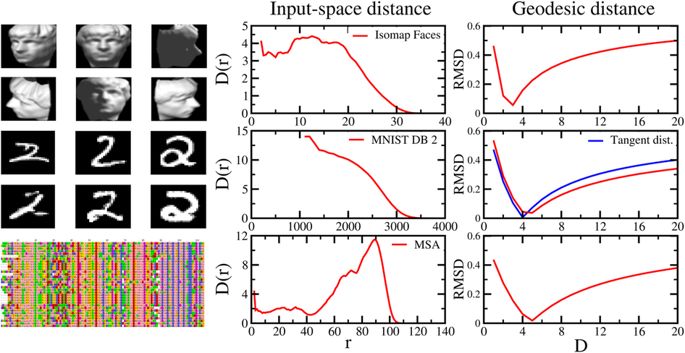
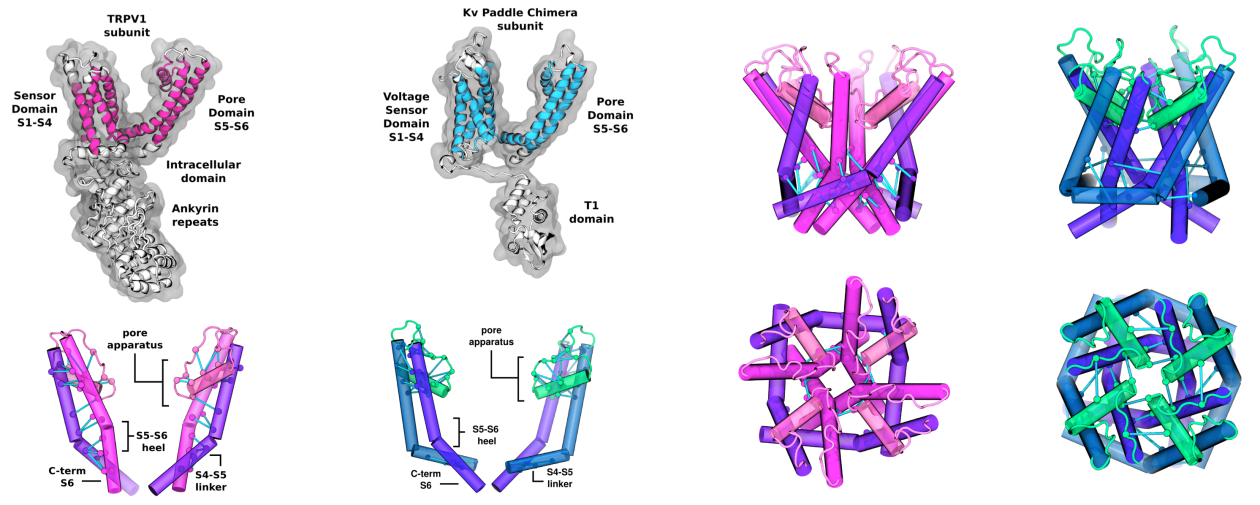
Patterns of interacting amino acids are so preserved within protein families that the sole analysis of evolutionary comutations can identify pairs of contacting residues. It is also known that evolution conserves functional dynamics, i.e., the concerted motion or displacement of large protein regions or domains. Is it, therefore, possible to use a pure sequence-based analysis to identify these dynamical domains? To address this question, we introduce here a general coevolutionary coupling analysis strategy and apply it to a curated sequence database of hundreds of protein families. For most families, the sequence-based method partitions amino acids into a few clusters. When viewed in the context of the native structure, these clusters have the signature characteristics of viable protein domains: They are spatially separated but individually compact. They have a direct functional bearing too, as shown for various reference cases. We conclude that even large-scale structural and functionally related properties can be recovered from inference methods applied to evolutionary-related sequences.